

Por Elizabeth Tapia*
Las tecnologías de secuenciación permiten “leer” la información almacenada en el genoma de cualquier especie, información que desde el punto de visto informático ha sido escrita en un alfabeto de cuatro letras ATCG (Adenina, Timina, Citosina y Guanina). Entre las potenciales aplicaciones de las nuevas tecnologías de secuenciación de tercera y cuarta generación, se encuentra el desarrollo de la medicina personalizada. Sin embargo, los costos de estas herramientas son todavía demasiado elevados para que pueda utilizarse masivamente.
En el grupo de Bioinformática del Centro Franco Argentino de Ciencias de la Información y de Sistemas (CIFASIS-CONICET/UNR) trabajamos en métodos para hacer más económica esta tecnología a través de la secuenciación paralela de cientos o miles de muestras de ADN.
Entre las potenciales aplicaciones de las nuevas tecnologías de secuenciación de tercera y cuarta generación, se encuentra el desarrollo de la medicina personalizada
A partir de muestras biológicas con cantidades ínfimas de ADN, los secuenciadores generan archivos de datos que contienen lecturas de la información genómica. Estas lecturas se realizan casi de forma aleatoria de a fragmentos de un tamaño promedio conocido según la tecnología.
Sin embargo, una variable importante a tener en cuenta es que los secuenciadores son propensos a errores: en las lecturas pueden sustituir una base por otra, borrar o insertar una base que no corresponde. En la actualidad, cuando se secuencia un genoma completo o zonas determinadas se hace a través de lecturas cortas, esto implica que se lee toda la información muchas veces en trozos pequeños de aproximadamente 300 bases. Como los errores son aleatorios y generalmente de sustitución de bases, al repetir varias veces el proceso, se pueden corregir a través de un consenso por agrupamiento de lecturas similares.
Las nuevas tecnologías de secuenciación de tercera y cuarta generación (3/4G) ofrecen lecturas de hasta 30 mil bases, 100 veces más que las de segunda generación de lectura corta limitadas a 300 bases. De esta manera, se puede “leer” de forma continua regiones genómicas con más de un gen e, incluso, pequeños genomas completos.
La posibilidad de lecturas largas es importante para el desarrollo de la medicina personalizada. Para cada tipo de enfermedad podrían considerarse regiones genómicas de interés y ser analizadas para cada individuo de modo de ayudar en la toma de decisión médica sobre diferentes terapias. En la actualidad, las terapias se diseñan para el promedio de los individuos, lo cual explica la ineficacia y a veces su efecto contraproducente en ciertos grupos.
Para cada tipo de enfermedad podrían considerarse regiones genómicas de interés y ser analizadas para cada individuo de modo de ayudar en la toma de decisión médica sobre diferentes terapias.
Para acercar este futuro es necesario reducir los costos y tiempos de secuenciación de las tecnologías 3/4G. Este objetivo puede alcanzarse con la secuenciación de muchas muestras en paralelo, por ejemplo, muestras de diversos individuos o variedades vegetales. Cuantas más muestras puedan ser analizadas simultáneamente, mayor será la reducción del costo de secuenciación por cada una. Para ello, se emplea una técnica conocida como multiplex, que permite el marcado de muestras biológicas mediante códigos de barras de ADN, secuencias conocidas de una longitud fija de las bases Adenina, Timina, Guanina y Citosina (ATCG) que se pueden sintetizar en el laboratorio.
Los códigos de barra de ADN, como los de los productos en el supermercado, deben ser diferentes entre sí, tanto más diferentes cuanta más posibilidad exista de errores en su lectura. El secuenciador lee la información genómica de las muestras sin importar su origen y lee en particular la secuencia del código de barra de ADN. Estas lecturas de códigos nos permiten revertir el proceso de multiplexación, recuperando la información genómica para muestras individuales. Si la lectura de un código es errónea debido a los errores del secuenciador, podría asignarse a una muestra equivocada. La incidencia de esta fuente de error en aplicaciones de diagnóstico médico debe ser controlada.
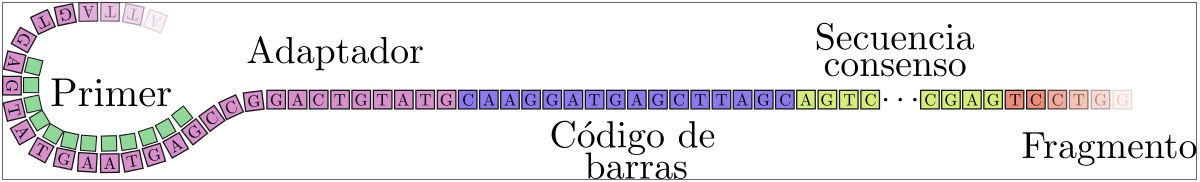
Las nuevas tecnologías de secuenciación de lectura larga presentan el desafío de tasas de error marcadamente superiores a las de lectura corta, en el rango del 11-15% con un componente importante de errores de inserción y borrado de bases. Estas características limitan fuertemente el diseño de códigos de barra de ADN lo suficientemente robustos como para reducir los errores de asignación incorrecta de lecturas a muestras a un nivel compatible con aplicaciones médicas.
Para abordar esta dificultad, desde el CIFASIS ideamos un método sistemático para el diseño de las secuencias de nucleótidos que pueden usarse como códigos de barra de ADN capaz de resistir los niveles de error mencionados. La lectura de un código de barra puede ser errónea, pero aun así puede procesarse mediante informática para recuperar su identidad original evitando, de esta manera, la eventual asignación incorrecta de lecturas a muestras. Este trabajo fue publicado recientemente en la revista internacional Bioinformatics.
Estos códigos de barra prometen un desempeño muy superior a los métodos existentes para las tecnologías de tercera generación que se están comenzando a usar. Su diseño es completamente flexible, dependiendo de la aplicación, se puede especificar cuantas muestras se analizarán y cuantos errores se prevén en la asignación de lecturas a muestras y a partir de ello diseñar un conjunto de códigos de barra de ADN con la longitud óptima de letras ATCG para satisfacer los requerimientos de una aplicación particular. Este factor es relevante para reducir los costos de los kits de multiplexado.
Artículo:
Joaquín Ezpeleta, Flavia J. Krsticevic, Pilar Bulacio, and Elizabeth Tapia. Designing robust watermark barcodes for multiplex long-read sequencing. Bioinformatics first published online June 3, 2016 doi:10.1093/bioinformatics/btw322
* Elizabeth Tapia directora del grupo de Bioinformática del Centro Franco Argentino de Ciencias de la Información y de Sistemas (CIFASIS-CONICET/UNR) y profesora asociada de la Facultad de Ciencias Exactas, Ingeniería y Agrimensura de la UNR.




